Site Map
Download
Pre-built target (.BED files)
Pre-built exclusion feature (.BED files)
Pre-built microarray (.BED files)
Publication
- Manuscript is currently under review.
|
AnyExpress an integrated toolkit for cross-platform gene expression data |
 |
Manual
How to create a .file to be used in Anyexpress$ cd /your_path/closedPlatform_AffyHG133PLUS2 Run bowtie to align microarray probe sequence (.fasta file) against human genome and save the output as .bowtie file $ /your_path/bowtie-0.12.5/bowtie -t -n 0 /bmi-data/bowtie-0.12.5/indexes/human/hg19 -f Affymetrix_HGU133PLUS2.fasta Affymetrix_HGU133PLUS2.bowtie Process the bowtie output (.bowtie) into .BED format for anyexpress, select five columns( chromosome, start position, end position, probe identifier and strand) and reagrrange columns to create .BED format file.As bowtie does not gives an end position, it has to be calcuated as: end position = start position + length of probe sequence - 1 (1) Linux $ awk '{ FS="\t"; OFS="\t"; print $3, $4, $4+length($5)-1, $1, $2 }' Affymetrix_HGU133PLUS2.bowtie > Affymetrix_HGU133PLUS2.BED & (2)Window (awk.exe download) C:\ awk Affymetrix_HGU133PLUS2.bowtie Affymetrix_HGU133PLUS2.BED $ cd /your_path/openPlatform_Illumina_GA Run bowtie, an external alignment software, to .fastq file $ /your_path/bowtie-0.12.5/bowtie -t -n 0 /bmi-data/bowtie-0.12.5/indexes/human/hg19 -q SRR002320.fastq SRR002320.bowtieConvert bowtie output file into .BED format file. Extract, chrom, start, end, length, identifier and strand. 'end' is calculated as 'end' minus 'length' of the read sequence. (1)Linux $ awk 'BEGIN {FS= "\t"; OFS="\t"} {print $3, $4, $4+length($5)-1, $1, $2 }' SRR002320.bowtie > SRR002320.BED & (2)Window (awk.exe download) C:\ awk SRR002320.bowtie SRR002320.BED [Reference: Marioni et al., 2008] BindAffyCelBinds multiple Affymetrix microarray .cel files column-wise into a single probe-by-sample text file. $ anyexpress BindAffyCel /path_to_.CEL_files MarioniAffy_bound.txt 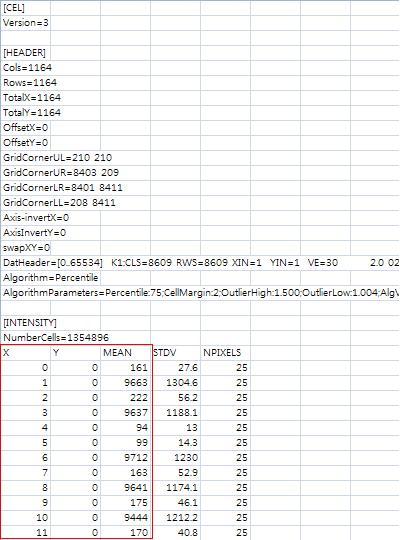

BuildExclusionFeatureCreates a files to be used for exclusion feature to filter out undesirable tags. For example, a probe containing a single SNP is undesirable one.Therefore, a user wants to eliminate it in a certain research context. Note: This tool can build more than one exclusion feature at a time. $ anyexpress BuildExclusionFeature /path_to/dbsnp129-single.BED /path_to/human_snp130_May2010.BED 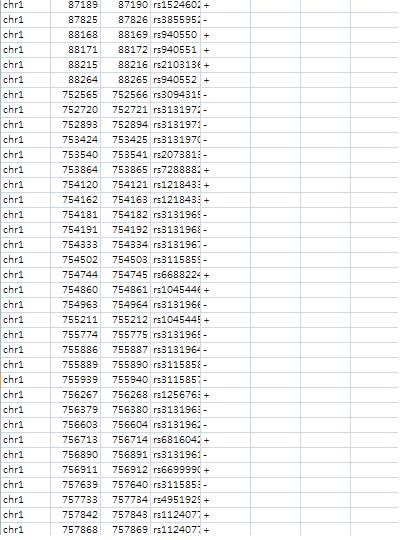
BuildTargetCreates a file to be used as a reference target against which tag position will be matched using the user-selected transcriptome database (.BED file). The target identifier must consist of two substrings concatenated by '@', i.e. targetID = 'superID' + '@' + 'subID'. For the 'BRCA1' gene, the identifier (with the corresponding target) could be represented for example as 'BRCA1@Exon2' (official gene symbol), 'NC_007294@Exon2' (RefSeq), or 'ENSG00000012048@Exon2' (Ensembl). $ anyexpress BuildTarget /path_to/human_refGene_2010May.BED 
CombineCombines both open- and closed-platform gene expression data into a single text target-by-sample text file. $ anyexpress Combine -c /path_to/Affymetrix_U133A.BED /path_to/Marioni_normalize.txt -o /path_to/SRR002323.BED -t human_refGene_2010May -e dbsnp129-single -p /path_to/projectName
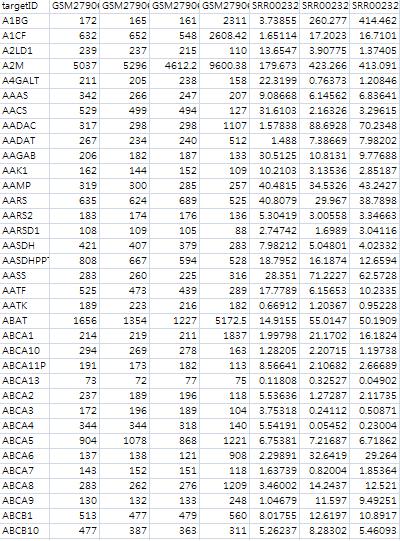
DisplaySysPrints currently available reference targets and exclusion features in the user's local directory $ anyexpress DisplaySys 
NormalizeColumnBoundSamplesPerforms quantile-normalization on a probe-by-sample text file. $ anyexpress NormalizeColumnBoundSamples Marioni_bound.txt Marioni_normalize.txt

PlotCreates a coverage plot along the genomic region (.bedGraph file) to be uploaded to the UCSC genome browser for viewing. $ anyexpress Plot projectName chrom strand start end 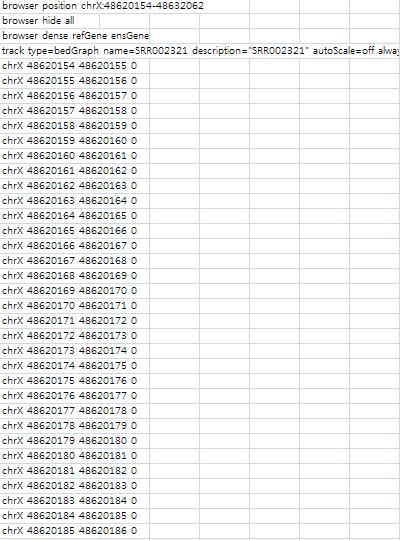
Memory allocationUsers can allocate memory size to any modules in anyexpress after -m. Default memory size is 2GB. If users want to allocate 4GB memory to Combine module, see the command below. $ anyexpress Combine -c /path_to/Affymetrix_U133A.BED /path_to/Marioni_normalize.txt -o /path_to/SRR002323.BED -t human_refGene_2010May -e dbsnp129-single -m 4 -p /path_to/projectName |