Site Map
Download
Pre-built target (.BED files)
Pre-built exclusion feature (.BED files)
Pre-built microarray (.BED files)
Publication
- Manuscript is currently under review.
|
AnyExpress an integrated toolkit for analysis of cross-platform gene expression data |
 |
Getting started
Install and configure1. Start the Bash shell
2. Download the anyexpress executables, unzip it to your local computer and obtain the current path to AnyExpress (to be used in step 4 below).
3. Open .bash_profile file
4. Add the following four lines to .bash_profile file
5. Reload the .bash_profile file
6. Verify the environment variable and the path setting
Get the example dataDownload the example data , unzip it and copy all the files to the directory ‘examples’.
Build a reference targetWe refer to a target as a biologically meaningful expression unit against which tags(=probes/reads) will be matched using their genomic positions. Each target is a collection of five attributes: chromosome, strand, start position, end position, and identifier. AnyExpress accepts the target as a .BED format file where those five fields are separated by tabs. A separate .BED file is needed per unique combination of followings:
For example, following four .BED files of human are considered to be different targets:
To build a target in anyexpress, issue the following command. $ anyexpress BuildTarget /path_to/human_refGene_2010May_gene.BEDYou can build multiple targets with a single run of BuildTarget: $ anyexpress BuildTarget /path_to/target_A.BED /path_to/target_B.BED /path_to/target_C.BEDEXERCISE: Type the command below in an examples directory.
Build an exclusion featureA exclusion feature is a biologically meaningful entity against which probe/reads will be compared to be removed. Exclusion features allow users to apply a filter against the tags to select out undesirable ones. Previous studies have shown the negative effect of low quality microarray probes on measurement of gene expression abundance and consequently on the interpretation of the results. For example, the presence of SNPs within the probe sequence would cause incorrect estimation of mRNA abundance.Therefore, a user wants to eliminate it in a certain research context. Note: This tool can build more than one exclusion feature at a time. To build an exclusion features in anyexpress, issue the following command. $ anyexpress BuildExclusionFeature /path_to/dbsnp129-single.BED You can build multiple exclusion features with a single run of BuildExclusionFeature : $ anyexpress BuildExclusionFeature /path_to/ex_feature_A.BED /path_to/ex_feature_B.BED /path_to/ex_feature_C.BEDEXERCISE: Type the command below in an examples directory.
Bind Affymetrix .cel filesWe defined the input format of closed-platform samples for AnyExpress as a single column-bound, tab-delimited text file where the first column is a probe identifier followed by measurement values of the samples in the second column. This is a common data format for micrroarray in non-Affymetrix platforms. However, in Affymetrix, each sample is a .cel file and they need to get bound column-wise before main analysis. AnyExpress provides a scalable binding tool, AnyExpress BindAffyCel, to create a single column-bound file from a large number of Affymetrix .cel File. To bind Affymetrix .CEL files in anyexpress, put all .cel files of the sample platform into a single direcotry and issue the following command. $anyexpress BindAffyCel /path_to_CEL_files output_bound.txt EXERCISE: Type the command below in an examples directory.
Normalize a probe-by-sample microarray fileQuantile-normalization is a rank-invariant transformation of measurement values to have the identical distribution of measurement values across all samples once they get processeds. The column-bound file can be directly used for an input to anyexpress Combine, but it is highly recommended to first perform between-sample normalization of this data to remove systematic bias and enable fair comparison among samples. To peform quantile-normalization to a probe-by-sample text file of the same microarray platform in anyexpress, issue the following command. $ anyexpress NormalizeColumnBoundSamples probe_by_sample.txt output.txt EXERCISE: Type the command below in an examples directory.
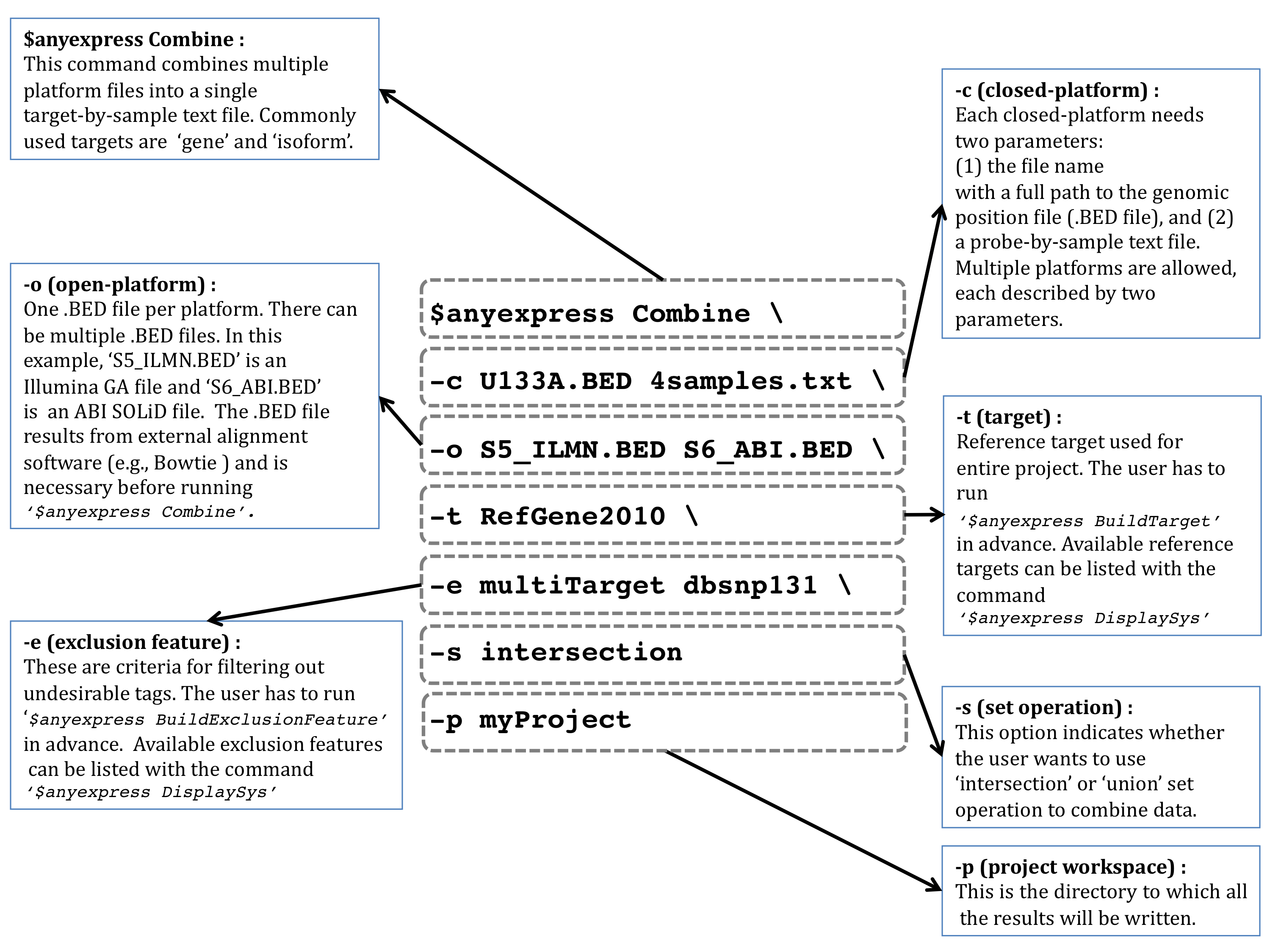
Combine cross-platform filesA summarization file is created per platform during the < Summarize > process, then the multiple summarization files are merged into a gene-by-sample text file through a < Join > process during < Combine >. To combine both open- and closed-platform gene expression data, issue the following command. $ anyexpress Combine -c /path_to/U133A.BED /path_to/4samples.txt -o /path_to/SRR002322.BED -t human_refGene_2010May -e multiTarget dbsnp129-single -p /path_to/projectName A detailed instruction on Combine is illustrated in a figure below:
Type the command below in an examples directory.
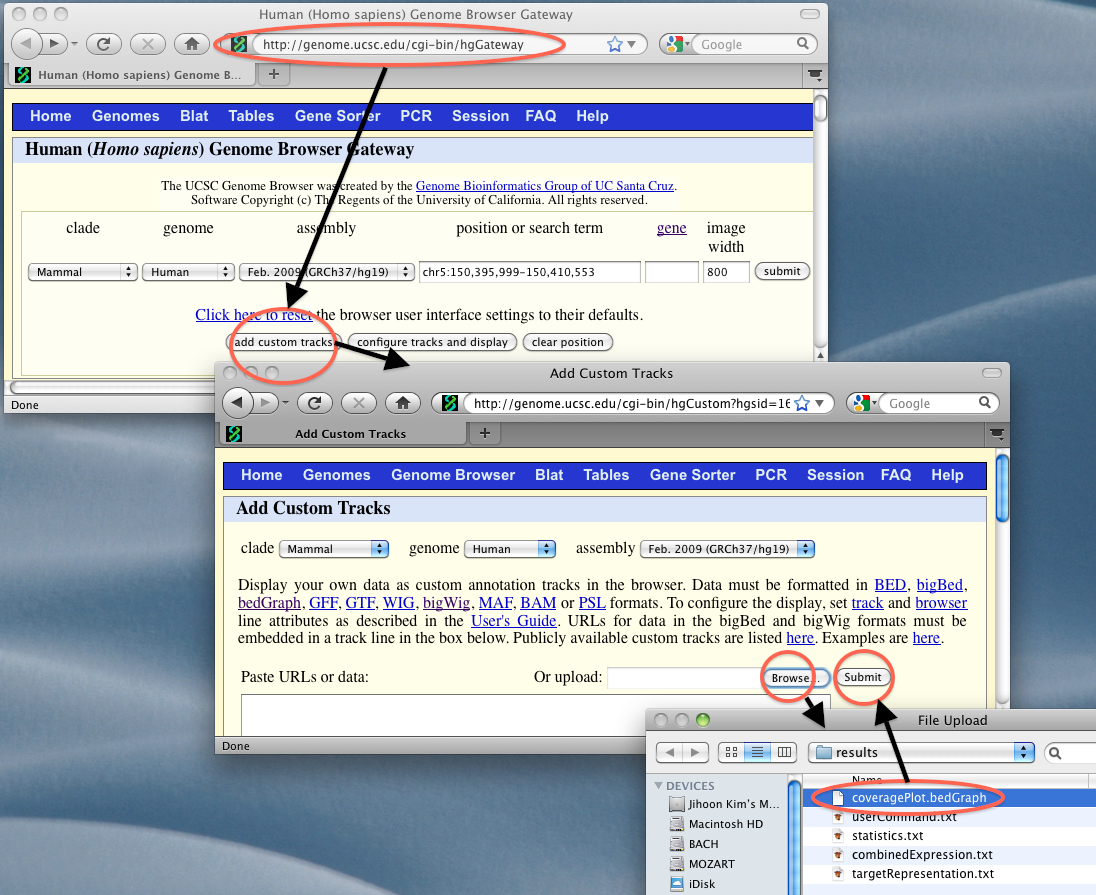
Draw a coverage plotThe user can draw a plot by typing in five parameters: a directory in which the users Project is, chromosome, strand (‘forward’ or ‘backward’), start position, and end position. Based on user’s combined results in Project, each open-platform and each sample in a closed-platform are drawn as tracks (rows) in the .bedGraph file. The user needs to upload this file onto the UCSC Genome Browser through his/her own web-browser. To draw a coverage plot (.bedGraph format), issue the following command. $ anyexpress Plot projectName chrom strand start end EXERCISE: Type the command below in an examples directory.
|